This blog is really just a place holder for my notes on using Git, which is probably the most widely used version control system. I swapped over from using CVS a while ago and, while I love Git, it can be a little tricky to get used to.
For my remote repositories I tend to use GitLab, because when I initially develop projects I like to keep them private, and GitLab offers free private accounts.
To start a new project you can either set up a repository on the website of your favourite remote repository (such as GitLab) and use ‘git clone’ to copy the repository to your local machine or you can create a repository locally (by moving to the directory where you store your files and using ‘git init’) and then push the repository (‘git push’) to copy it to the remote repository.
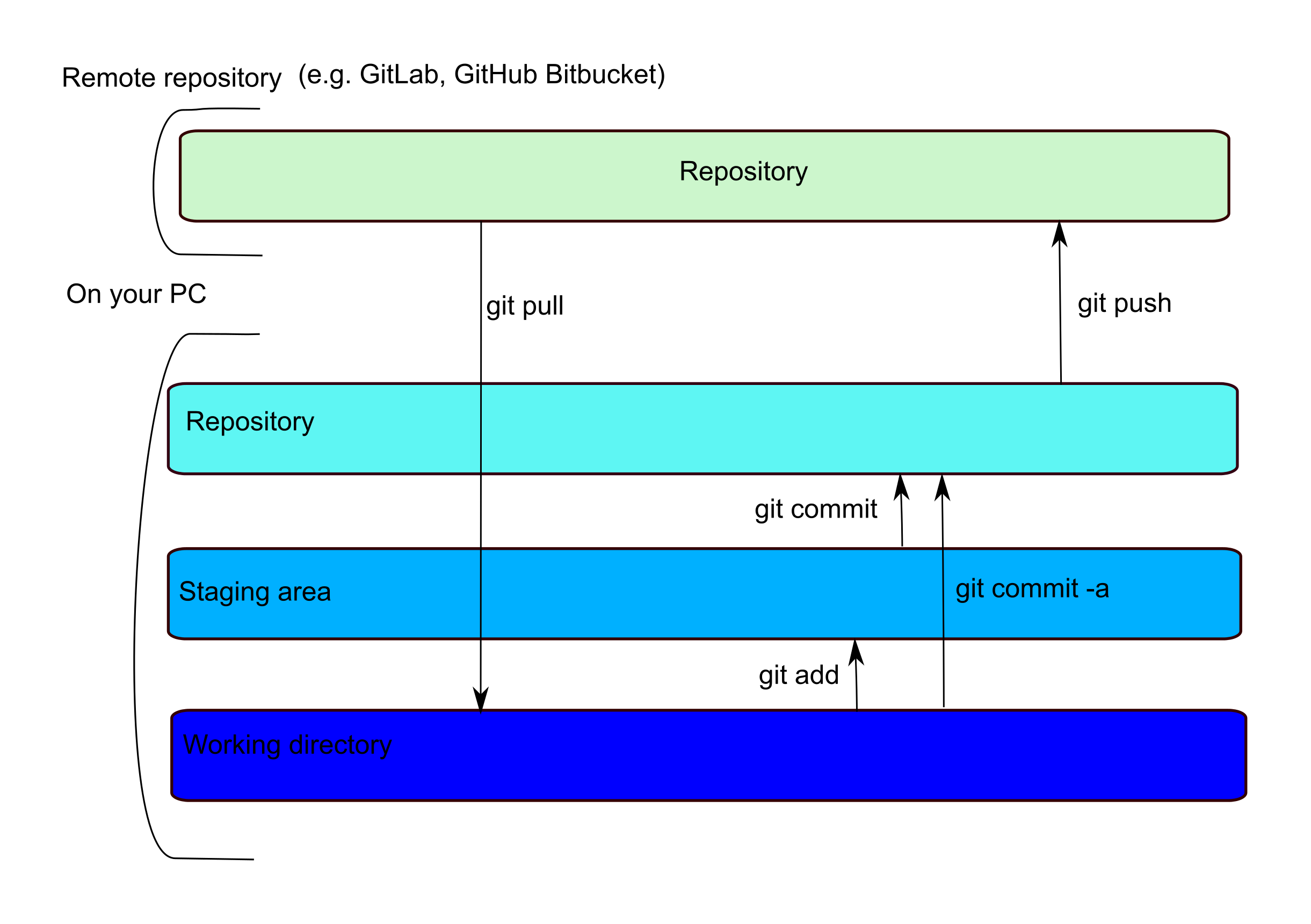
What can be a little confusing is that locally, while there is only one physical copy of your file, there are indexes that track if a file has been ‘added’ to a staging area or ‘committed’ to your local repository. This means that there is a two-step commit… you ‘git add’ to move a file to the staging area and ‘git commit’ to move it to the local repository. A ‘git push’ will move any committed files to your remote repository, where they can be accessed by collaborators.